Research
Overview
- In this page, I highlight my research on how machine learning and computational models enable understanding, mapping, and predicting cellular biology. The work is organized into four directions: (1) generative modeling of cellular responses to perturbations; (2) tissue architecture and ecosystem reprogramming with spatial genomics; (3) generative multimodal tissue foundation models; and (4) core machine learning methods for biomedical generative AI.
- Before 2018, as part of my AI program master thesis, I developed Deep Packet, the first neural network architecture for network traffic classification, and it has become one of the seminal papers in the field.
- Papers selected as cover:



- See a full list of papers on Google Scholar.
Generative modeling of cellular responses to perturbations
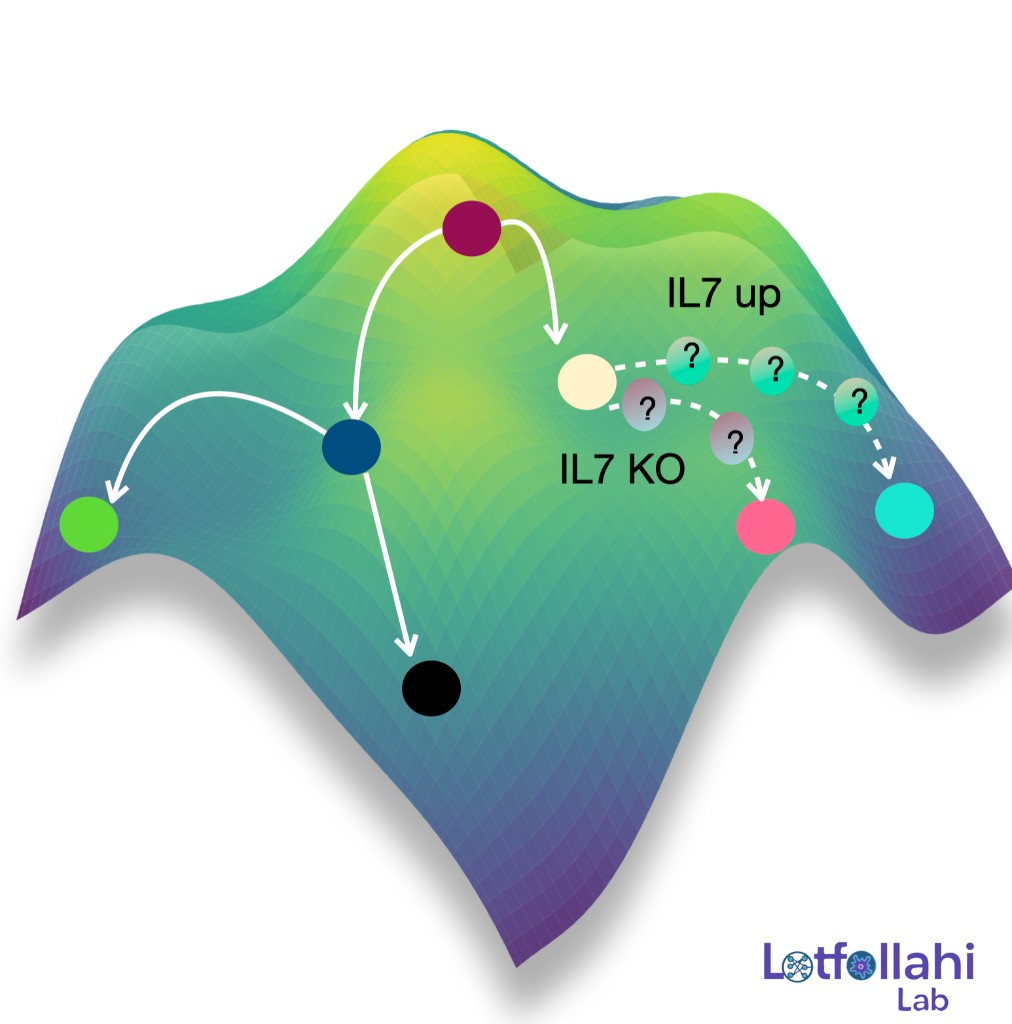
We develop generative AI models that predict how cells change in response to perturbations—such as drugs, CRISPR knockouts, disease context, or other interventions—enabling counterfactual biology: given a cell measured in one condition, what would it look like under another? This research direction has evolved into a modeling framework (some call it virtual cell modeling) aimed at learning reusable representations of cell state and using them to predict cellular responses to perturbations, including out-of-distribution generalization to unseen perturbations and combinations. The long-term goal is to turn perturbational datasets into a predictive engine for hypothesis generation, mechanism discovery, and prioritization in large-scale screens.
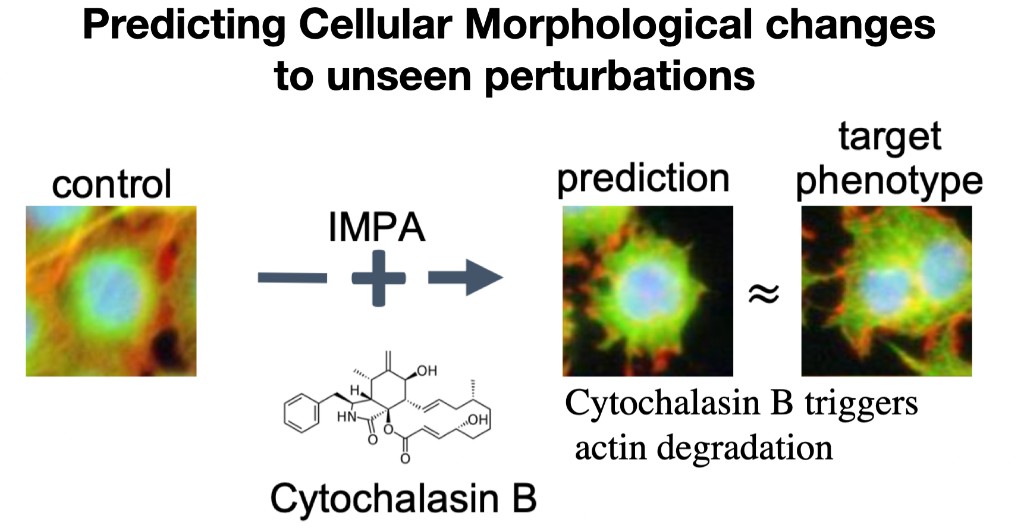
Across single-cell omics and high-content microscopy, we introduced a series of generative approaches that learn how perturbations transform cellular state and phenotype. Our early work scGen models perturbation effects with simple latent-space arithmetic. We then developed trVAE, which reframes perturbation prediction as distribution matching—moving cells from a control distribution to a perturbed condition. Later, during my time at Facebook AI, we introduced the Compositional Perturbation Autoencoder (CPA) to predict combinatorial perturbations such as drug combinations or double CRISPR knockouts, and extended it to handle unseen drugs (chemCPA). In parallel, we developed IMPA (Image Perturbation Autoencoder) to predict perturbation-induced morphological responses in high-content microscopy using untreated cells as input, addressing the challenge that only a small fraction of perturbations show measurable activity in experimental screens. More recently, we introduced CellDISECT, where we introduce multiple counterfactuals for disentangling covariates and predicting cellular responses.
Selected papers:
- scGen — Nature Methods (2019) — [paper], [code], [press]
- trVAE — Bioinformatics (2020) — [paper], [code], [talk]
- CPA — Molecular Systems Biology (2023) — [paper], [code], [blogpost]
- chemCPA — NeurIPS (2022) — [paper], [code]
- IMPA — Nature Communications (2025) — [paper], [code]
- CellDISECT — bioRxiv (2025) — [paper], [code]
Tissue architecture and ecosystem reprogramming with spatial genomics
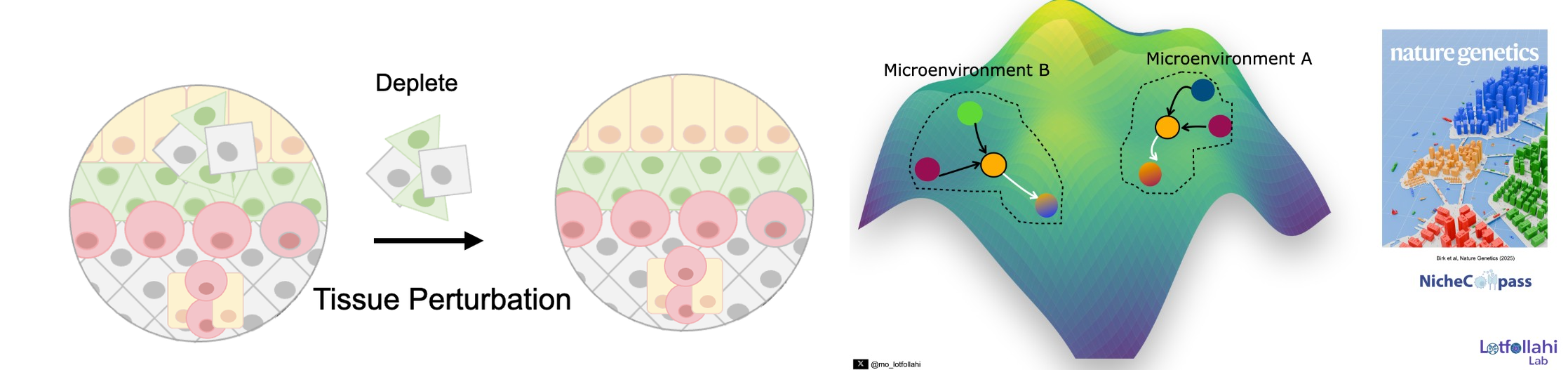
Spatial genomics makes it possible to study tissue organization at cellular resolution, but two fundamental challenges remain. First, can we identify tissue microenvironments (niches) and quantitatively define what makes them distinct—based on the cellular pathways and communication programs that structure them? Second, can we move beyond description and simulate perturbations of tissue microenvironments to predict how local ecosystems and cellular states reprogram in response to interventions?
To address the first challenge, we developed NicheCompass, a pathway-informed graph deep learning framework grounded in principles of cellular communication. NicheCompass learns interpretable representations that capture signaling and interaction programs, enabling robust niche discovery and quantitative characterization across spatial samples and disease contexts.
To address the second challenge, we developed MintFlow, a generative AI approach that learns how local tissue microenvironments imprint and reprogram cellular state, and enables in silico tissue perturbations—for example, depleting or replacing specific cell populations and predicting resulting responses at micro- and macro-scales. MintFlow shifts spatial analysis from descriptive mapping toward predictive simulation of tissue ecosystem reprogramming, supporting unbiased mechanism discovery and translational hypothesis generation across human disease settings.
Selected papers:
Generative Multimodal Tissue Foundation Models
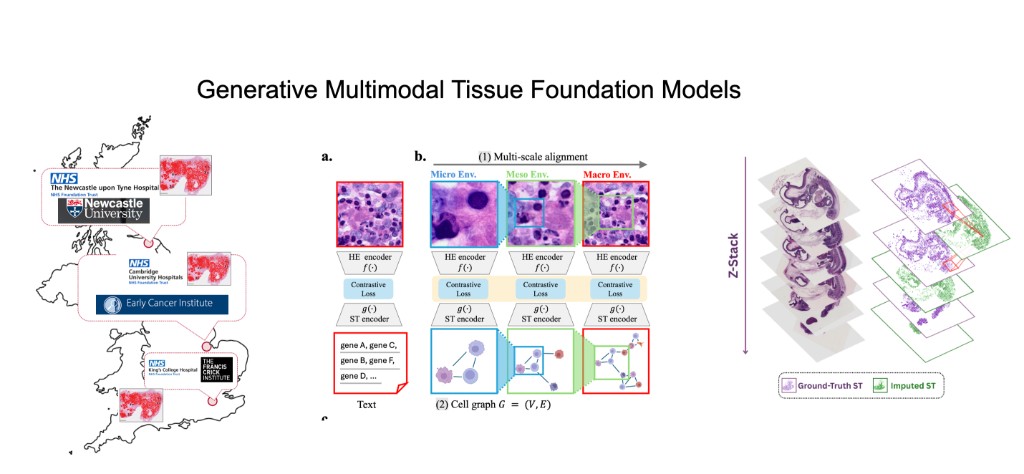
Our lab generates massive, patient-linked multimodal tissue datasets collected across hospitals in the UK and internationally—combining H&E whole-slide imaging, spatial transcriptomics, transcriptomics, proteomics, and DNA sequence. Our research direction is to build generative multimodal foundation models that link these modalities and enable counterfactual tissue biology: given a tissue sample measured in one modality or context, generate what it would look like in another modality (with uncertainty), while preserving spatial organization, tissue architecture, and cell–cell interactions. A central objective is "cheap → expensive" generation, e.g., predicting spatial transcriptomics from routine histology, and ultimately learning patient-level tissue representations that generalize across sites, protocols, and cohorts.
Building on HoloTea, we move beyond 2D slice prediction to 3D virtual tissues by generating spot-level gene expression from H&E using a 3D-aware flow-matching framework that leverages adjacent sections for volumetric continuity and incorporates biologically appropriate priors for count data.
We then extend this into robust, reusable representations with SIGMMA, which learns hierarchical multi-scale aligned embeddings of H&E and spatial transcriptomics, representing spatial expression as a cell graph to preserve tissue topology and interactions across micro/meso/macro scales.
Together, these advances define a roadmap toward tissue foundation models that can generate missing modalities, assemble coherent 3D molecular anatomy, and enable scalable biomarker discovery and mechanistic hypothesis generation in real-world clinical tissue data.
Selected papers:
Core Machine Learning Research
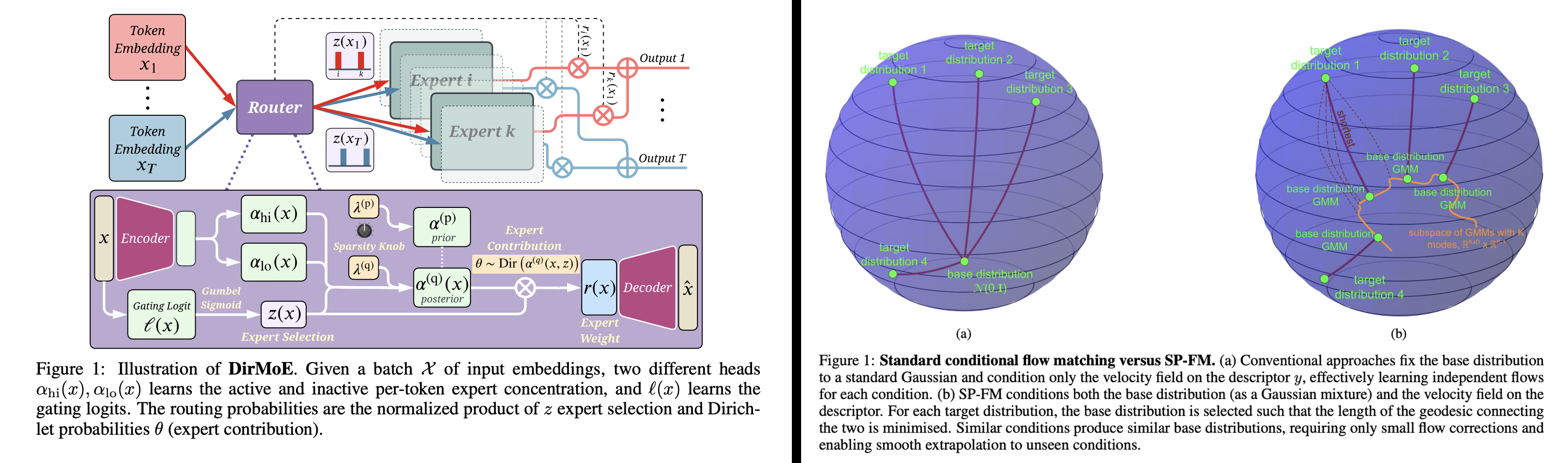
To answer hard biological and clinical questions with generative AI, we can't simply port existing architectures from vision or language. Biomedical data is heterogeneous, noisy, missing by design, and heavily shifted across cohorts—so we often need new architectures, new probabilistic/mathematical modeling choices, and new training objectives to make generative models scalable, stable, and scientifically meaningful. This is a core direction of our lab: we develop foundational ML methods for generative modeling that later power our multimodal tissue and patient models.
Two recent mixture-focused examples (ICLR 2026) illustrate this philosophy. DirMoE introduces a fully differentiable Mixture-of-Experts router that disentangles which experts activate (Bernoulli "spike") from how probability mass is allocated among them (Dirichlet "slab"), enabling end-to-end training with interpretable control over sparsity and specialization.
In a complementary direction, Shortest-Path Flow Matching (SP-FM) improves conditional generative modeling by conditioning both the base distribution (as a Gaussian mixture) and the velocity field on the condition, effectively choosing a base that shortens the path to the target distribution and strengthening extrapolation to unseen conditions.
Selected papers:
Machine learning to build single-cell atlases

Our lab has pioneered machine learning methods that make single-cell atlases reusable, extendable references—so new datasets can be mapped, annotated, and compared in a shared coordinate system rather than re-integrated from scratch.
We introduced scArches for reference mapping by transfer learning, enabling query datasets to be mapped into a pretrained atlas without retraining the reference. We extended this to evolving cell identity structure with treeArches (updating cell-type hierarchies). We expanded atlas building beyond RNA with multimodal generative models: Multigrate integrates partially overlapping modalities (RNA/ATAC/protein) into unified multimodal references, and mvTCR integrates TCR sequences + scRNA-seq for immune atlases. To improve biological meaning and scale, we developed expiMap for interpretable gene-program embeddings and scPoli for population-level references that learn sample- and cell-level structure and provide uncertainty-aware annotation of new data.
Community impact (examples)
The community has extensively leveraged these approaches. Here are examples that our lab contributed to, showcased in building the first integrated reference atlas for lung, and more recently integrated spatial atlases for fibroblasts and skin.